Human Intestinal Microbiota in Response to Antibiotics, Nicotine and Caffeine.
Undergraduate Research Topic: Maximiliano Rodriguez.
2017-2019
Abstract
Intestinal microbiota play a number of critical roles in the health of the organism.
They assist in nutrient metabolism and play a key role in the host’s immune
response. Current research is revealing that there may be a significant impact from
intestinal bacteria on the gut-brain axis and that they play a downstream role in
several neurological disorders. Here we present data from a 26-day single-person
pilot study where we looked at the effects from the use of tobacco products on the
microbial diversity during an antibiotic treatment and a 2-week recovery period
following the antibiotic course. During the course of the project detailed dietary
and behavioral data were collected and later used to construct an environmental
matrix for multivariate analysis. The goal of this project was to see how
behavioral factors influence both the antibiotic driving decline of gut microbiota
as well as the recovery period following an antibiotic course.
Research Article

Analysis Of Microbiome In Various Human Medical Conditions\nUsing Next-Generation
Sequencing Of The 16S Rrna Gene.
Graduate Research Topic: Pallavi Sharma.
2022-2024
Abstract
Studies of human microbiomics, spanning diverse anatomical sites, offer a
unique perspective into the complex microbial communities within the body.
Influenced by age, gender, and disease, it mirrors broader microbial ecosystems.
This study specifically delves into the ocular and gastrointestinal tract
microbiome, comparing its composition in 43 healthy and diseased patients using
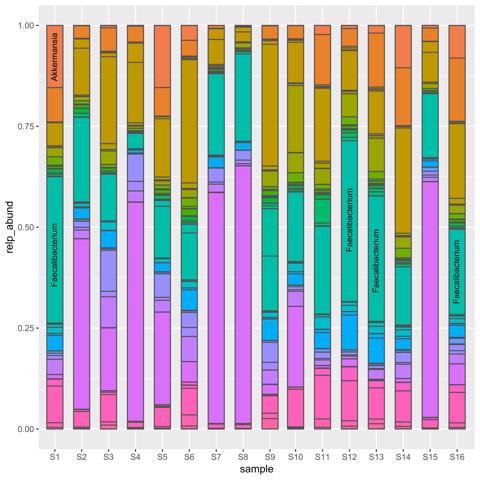
16S rRNA gene sequencing. We identified a rich microbial landscape, identifying
over 338 bacterial taxa. Notably, Acinetobacter, Cutibacterium, Acidovorax and
Herbaspirillum were prevalent in the ocular samples and Akkermansia,
Faecalibacterium, and Anaerostipes were mostly prevalent in gut samples.
Moreover, dry eye disease conditions were characterized by a higher prevalence
of Carnobacteriaceae, Acinetobacter, and Enterococcaceae, whereas healthy
eyes exhibited a distinct microbial composition, including the presence of genera
Streptococcus and Staphylococcus. The taxa richness in fecal samples from
patients with different disease conditions was strongly associated with an
increased abundance of the Propionibacteriaceae family in those with lower
gastrointestinal bleeding. The most common taxa abundant throughout the stool
samples were Anaerobutyricum, Lachnospiraceae, and Eubacteriales. These
specific microbiome signatures offer nuanced insights into ocular and gut health.
This integrated approach enhances our capacity to understand gut-eye
association and develop targeted therapeutic interventions, emphasizing the
translational potential of microbiomics in improving human health.
Thesis

Characterization of the Cervicovaginal Microbiota of Asymptomatic Women Using 16S rRNA
Gene Sequencing.
Graduate Research Topic: Daisy Ngetich.
2024-current
Abstract
The human microbiome, is a complex community of microorganisms residing in various body
sites, plays a crucial role in maintaining health and influencing disease outcomes.
Traditionally,
research has focused on understanding the microbiome in the context of medical
conditions,
often examining symptomatic patients to identify microbial imbalances associated with
diseases.
However, this research takes a novel approach by investigating the cervicovaginal
microbiome
in asymptomatic women, aiming to establish a baseline understanding of what constitutes
a healthy microbiome , predominantly featuring Lactobacillus species.
The study will examine how demographic factors such menstrual cycle phases and body mass
index (BMI), influence the cervicovaginal microbiome. The hypothesis posits that
significant
differences in microbiome composition exist across these variables, with specific
bacterial taxa
linked to positive reproductive health outcomes. This approach not only enhances our
understanding of a healthy microbiome but also lays the groundwork for future research
on its
implications for women's health.
Thesis

Rotavirus Effect on Gut Microbiome of Various Organisms.
Undergraduate Research Topic: Husna Chaudhary.
2024-current
Abstract
Human rotavirus (RV) is one of the world’s leading causes of death in children under the
age
of five (Harris et al., 2017). This epidemic has become a major issue globally in the
past few
decades, the worst of cases including African, Oceanic, and South Asian countries. RV
infects
the small intestine, a central location for beneficial gut bacteria often called the gut
microbiome
and can cause acute diarrhea that is fatal in young children (Iturriza-Gómara and
Cunliffe,
2017). Over the past several years, the microbiome has been studied extensively in its
independent state and how it is affected by diverse types of infection and
macromolecules via
diet. RV is one such case that temporarily alters the condition of the microbiota,
causing the
most severe symptoms, such as excessive vomiting, watery diarrhea, dehydration, etc.
(Bernstein, 2009). Through several different studies, discoveries were made on the
specifics of
the RV infection mechanism through multiple organisms, including mice, calves, piglets,
and
humans. Each of these organisms has key similarities and differences in their
microbiomes
and immune responses, which also fluctuate based on the concentrations of macromolecules
in the diet. (Kumar et al, 2022). By analyzing the overlapping characteristics, we can
distinguish properties repeatable between organisms. Carrying these discoveries to the
next
study allows us to determine which factors can decrease the effects of RV on the
microbiome
and if these correlations can be applied to humans. Changes in diet can strengthen the
microbiota, making them more resistant to viral changes. (Kashtanova et al, 2016). While
there is currently no effective cure for the human rotavirus (Bernstein, 2009), through
studies
like these, it may be possible to alleviate some symptoms or increase the chances of
survival
for the infants affected with this information.
Manuscript